Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, в основе которых лежат нарушения числа или структуры хромосом, то есть избыток или нехватка генетического материала, локализованного в той или иной хромосоме.
В норме у человека число хромосом равно 46, из которых 23 ребенок получает от матери и 23 аналогичные хромосомы от отца. В этом наборе гентического материала есть 2 особые хромосомы, которые были названы «половыми». Они определяют пол ребенка и ряд других важных признаков.
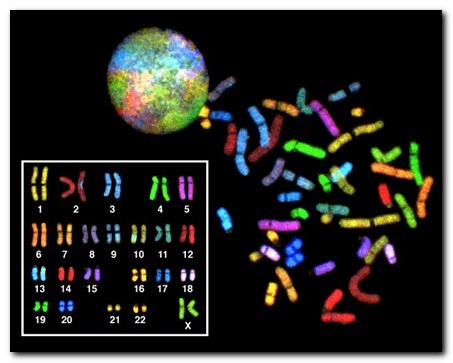
Таким образом, изменения числа хромосом (больше или меньше 46), а также изменение структуры хромосом (например, выпадение или удвоение даже небольшого кусочка хромосомы) получили название «хромосомные мутации».
Наиболее часто из них встречаются изменения модального числа хромосом — это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т.д.).
Число возможных изменений структуры хромосомы неисчислимое множество. К примеру, транслокации (обмен сегментами между разными хромосомами), делеции (выпадение участка хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и т.д.
Хромосомные мутации, возникшие в половых клетках (сперматозоидах или яйцеклетках) или на первых этапах деления клеток зародыша, как правило, передаются большинству клеток развивающегося организма, вызывая множественные аномалии развития, а многие хромосомные изменения плода могут стать причиной спонтанных абортов и выкидышей, что важно учитывать в семьях, воспитывающих детей с задержками развития.
К факторам риска, способствующим их возникновению, относят ионизирующую радиацию, инфекции и интоксикации матери, эндокринные нарушения, психические травмы, воздействие ряда лекарственных препаратов и некоторых физиотерапевтических методов лечения.
Наиболее точно установлено, что причиной появления ребенка с хромосомными мутациями является не молодой возраст матерей (свыше 40 лет).
В последнее время очень большое значение придается факту скрытого носительства хромосомных нарушений у родителей родившегося ребенка (сбалансированные транслокации, мозаицизм). Изучение данного вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичной формой заболевания.
Различают хромосомные синдромы, обусловленные изменением половых хромосом, и синдромы, вызванные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями аутосомных аномалий являются признаки психического и физического недоразвития, дисплазии (неправильное развитие), врожденные пороки развития (аномалии) и умственная отсталость различной степени тяжести. К врожденным порокам можно отнести: аномалии развития сердца, удвоение почки, расщелина неба, особенности строения кистей и стоп и многие другие. При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
Различные хромосомные синдромы встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых из них среди новорожденных следующая:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600
Не смотря на казалось бы не частую встречемость каждого отдельного синдрома, в целом хромосомные болезни у новорожденных наблюдаются не редко - с частотой около 1 : 100. Ежегодно в России рождается свыше 30 тыс. детей с хромосомной патологией. Спонтанные выкидыши являются результатом хромосомной патологии в более чем 50%.
Рассмотрим основные клинические проявления отдельных хромосомных синдромов, сопровождающихся умственной отсталостью и задержками психо-речевого развития.

Синдром Дауна - врожденное заболевание, характеризующееся умственной отсталостью и рядом признаков эндокринной недостаточности.
Синдром впервые описан английским врачом Дауном в 1866 г. Встречается с частотой 1 на 500 новорожденных. Частота встречаемсоти у мальчиков и девочек одинакова. В основе заболевания лежит аномалия хромосомного набора (47 вместо 46). Лишняя хромосома обнаруживается в 21 паре, в связи с чем этот синдром иногда называют «трисомией по 21-й хромосоме» (47, 21+). Выявлена связь частоты рождения больных с увеличением возраста матери. Приблизительно в 3—4% случаев отмечаются транслокационные формы синдрома Дауна, при которых общее число хромосом в кариотипе нормальное - 46, а дополнительная 21-я хромосома транслоцирована (присоединена) на другую аутосому. Это является результатом того, что один из фенотипически здоровых родителей является скрытым носителем сбалансированной транслокации. Именно за счет этих форм повышается риск повторного рождения больного ребенка у молодых матерей. Еще 3-4% случаев синдрома Дауна составляют мозаичные варианты, при которых в организме одновременно обнаруживают и трисомные, и нормальные клетки. Порой, при небольшом проценте трисомных клеток ребенок с ЗПРР внешне может выглядеть абсолютно нормальным.
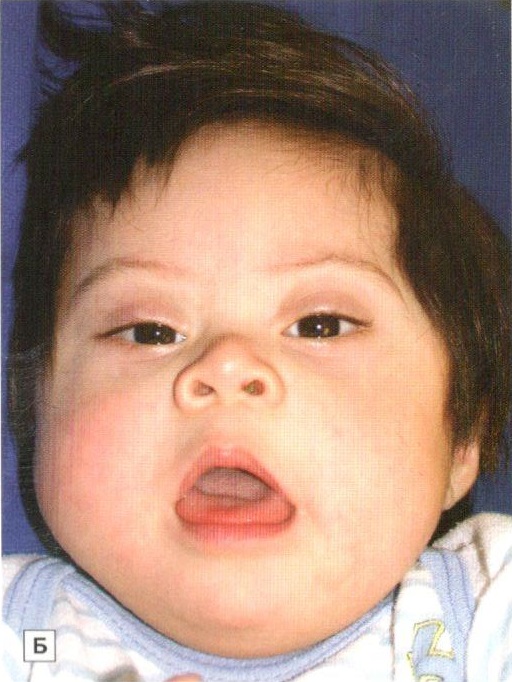
Установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также аномалии развития мозга и мозговых сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клиническая картина синдрома Дауна характеризуется проявлениями симптомов умственной отсталости. Характерен также и внешний вид таких больных: косо расположенные глазные щели, широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз, высокое стояние твердого неба (признаки эмбриональной задержки в развитии лицевого скелета), полуоткрытый рот, увеличенный высунутый язык с выраженными сосочками и глубокими бороздами (признаки дисфункции щитовидной железы), выпадение волос (дисфункция надпочечников), низкий рост, короткая шея, укороченные кисти и стопы, искривление мизинца, на ладонях имеется поперечная складка, на стопах увеличен промежуток между 1 и 2 пальцами, выражены внешние проявления гипогенитализма.
Такие дети с рождения отстают в росте, начинают поздно держать голову, сидеть и ходить. Речь, как правило, невнятная, словарный запас беден, произношение с дефектами в связи с недоразвитием высших мозговых функций, с одной стороны, и анатомическими аномалиями ротовой полости - с другой.
В клинической картине заболевания доминируют симптомы неврологической патологии, диффузная мышечная гипотония (снижение мышечного тонуса), благодаря чему больные гибки и иногда могут складываться как «перочинный ножик», расстройства координации движений, косоглазие, выраженные вегетососудистые нарушения.
Особенностью психического дефекта является относительная сохранность эмоциональной сферы по сравнению с тяжестью интеллектуального недоразвития. Так, больные ласковы, добродушны, послушны. Характерной особенностью таких детей является повышенная внушаемость, что является положительным фактором при проведении коррекционной работы и отрицательным при их развитии.
Уровень социального развития больных с синдромом Дауна зависит от степени и формы заболевания. Так, дети с более легкими формами умственной отсталости, хотя и медленно, но развиваютя, приобретая определенные навыки, знания, осваивая программу нескольких классов вспомогательной школы. Однако, как правило, большинство из них не достигают удовлетворительного уровня социальной адаптации и нуждаются в постоянной опеке. Им может быть оформлена инвалидность детства с момента точной диагностики заболевания. Особенностью возрастной динамики синдрома Дауна является позднее половое созревание и раннее появление признаков инволюции (25—30 лет). Мужчины с синдромом Дауна бесплодны, женщины могут давать потомство, половина которого также страдает синдромом Дауна.
Синдром Шерешевского—Тернера - симптомокомплекс проявлений врожденного, наследственно обусловленного недоразвития половых желез и передней доли гипофиза в сочетании с аномалиями соматического развития.
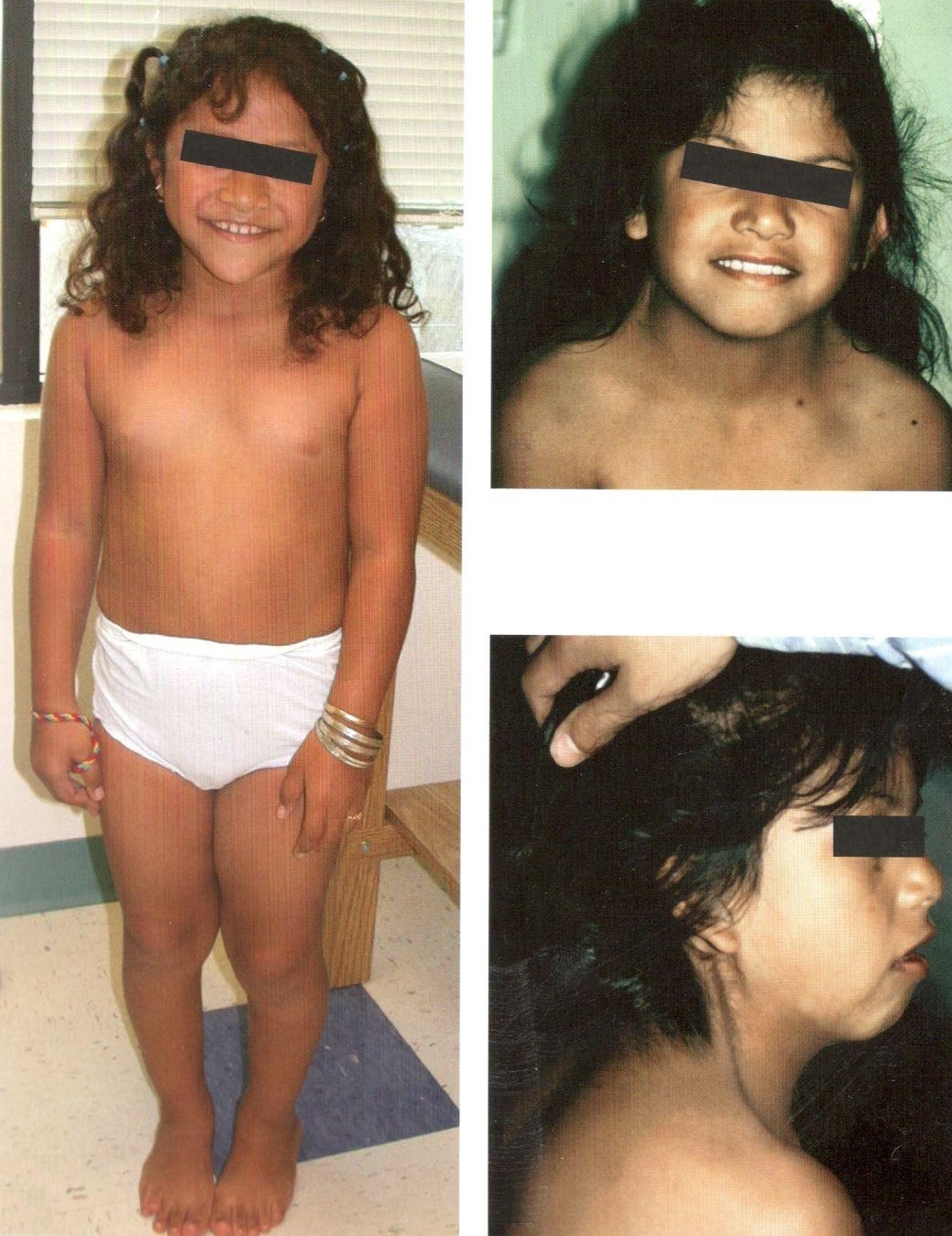
Впервые заболевание описано отечественным эндокринологом Н.А. Шерешевским (1925), а более подробно — американским эндокринологом Н. Тернером (N.H. Terner) л 1938 г. В основе заболевания лежит отсутствие одной хромосомы (половой Х-хромосомы) (45 вместо 46).
Клиническая картина синдрома характеризуется разной степенью умственной отсталости и ЗПРР, низким конечным ростом (135—145 см), замедлением полового развития, недоразвитием половых желез, аменореей, бесплодием и отсутствием грудных желез. Диспластические расстройства проявляются в виде короткой шеи и особых кожных складок, идущих от затылка к надплечью, укорочением 4 пальцев на руках и искривлением мизинцев, выраженной деформацией ушных раковин, наличием множественных пигментных родинок. Преимущественно данным синдромом страдают лица женского пола.
Синдром Клайнфелтера - заболевание, обусловленное нарушением числа половых хромосом (добавочные Х-хромосомы) (от 47 до 49), характеризующееся умственной отсталостью, нарушением смерматогенеза, недоразвитием яичек и вторичных половых признаков, а также нарушением пропорций тела. Впервые синдром описан американским эндокринологом Клайнфелтером (H.F. Klinfelter) в 1942 г. Его частота, по сводным данным, составляет до 2% среди умственно отсталых и до 0,5% (кадждый двухсотый мужчина) в среднем в мужской популяции.
Клинические проявления синдрома Клайнфельтера варьируют от внешне нормального и интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. Однако в ряде случаев уже в раннем возрасте у больных отмечаются характерные своеобразные симптомы физического развития: низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более отчетливо вышеперечисленные симптомы начинают обнаруживаться в подростковом, пубертатном возрасте. Характерен внешний вид взрослого больного с синдромом Клайнфельтера: высокий рост, астеническое сложение, узкие плечи, широкий таз, удлиненные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и оволосение по женскому типу, сутулость, выраженные евнухоидные пропорции и гинекомастия (набухание грудных желез). Постоянными признаками синдрома Клайнфельтера являются недоразвитие половых органов и бесплодие.
Степень интеллектуального недоразвития у больных выражена тем глубже, чем больше дополнительных половых хромосом обнаруживается в кариотипе (46 или 49). Так, умеренная умственная отсталость зачастую приближается к психическому инфантилизму, что клинически проявляется недостаточностью внимания, восприятия, памяти, абстрактного мышления, чрезмерной внушаемостью, подражательностью, подчиняемостью, несамостоятельностью, чрезмерной привязанностью к близким, нередко с элементом назойливости. Глубокая незрелость эмоционально-волевой сферы проявляется в виде повышенного настроения, с эйфорическим оттенком, склонностью к эксплозивным аффективным вспышкам, неспособностью к длительному волевому усилию и напряженной деятельности. У больных, как правило, отсутствуют чувство долга и ответственности. При легких формах заболевания больные осознают свою неполноценность, что приводит к внутреннему конфликту и возникновению у них невротических реакций. Данным синдромом страдают лица мужского пола.
Синдром ломкой Х-хромосомы (Fragile X syndrome, FraХ). Начиная с 1980 года большое значение придают синдрому ломкой Х-хромосомы (Хq27.3) – именно с ним связывают развитие более чем 50 наследственных расстройств, включая ранний детский аутизм и 30% случаев умственной отсталости у мальчиков. Хрупкий участок Х-хромосомы впервые обнаружил Labs (1969).
Полная мутация в Х-хромосоме возникает только у женщин, и происходит это в процессе гаметогенеза, поэтому почти всегда страдают мальчики, получившие единственную Х-хромосому от матери. У девочек, получивших вторую Х-хромосому от отца, также могут быть нарушения развития, но они менее выражены, а тяжелые патологии встречаются много реже, чем у мальчиков. В отдельных случаях девочки могут получить обе ломкие хромосомы от матери, в этом случае частота и тяжесть патологии будет одинаковой с мальчиками.
Клиническую триаду синдрома ломкой Х-хромосомы образуют:
1) умеренная до степени тяжелой умственная отсталость. Лишь 30% лиц мужского пола имеют интеллект, стремящийся к нижней границе нормы, а среди женщин – носительниц такой хромосомной патологии примерно у 30% обнаруживаются признаки умственного недоразвития;
2) характерные особенности строения лица и черепа: выдающийся вперед высокий лоб, прогнатизм и удлиненные уши;
3) мальчики имеют увеличенные в размерах тестикулы (макроорхидизм).
Наблюдаются, кроме того, эпилептические припадки, синдром гиперактивности с дефицитом внимания, у более чем половины мальчиков аутизм и подобные аутизму расстройства, различные нарушения развития речи, персеверации, эхолалия, другие отклонения.
Женщины, унаследовавшие ломкую Х-хромосому с полной мутацией от своих матерей, могут быть склонны к развитию атипической депрессии, а также шизофреноподобного заболевания.
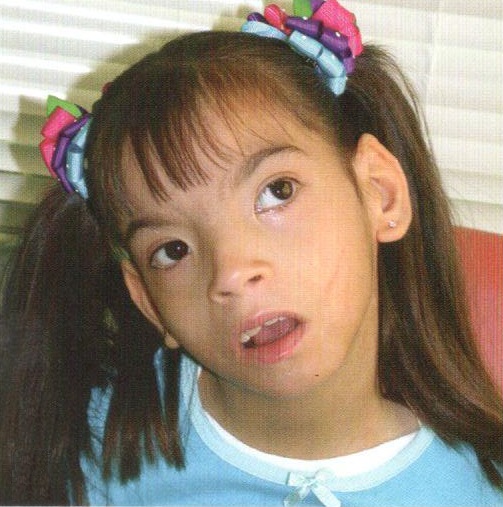
Синдром «кошачьего крика» - заболевание, обусловленное структурной аномалией 5-й пары хромосом (выпадение участка - делеция). Встречается преимущественно у девочек и характеризуется развитием умеренной или тяжелой умственной отсталости, задержкой физического развития и рядом диспластических признаков («антимонголоидный» разрез глаз, гипертелоризм, низкое расположение ушных раковин, поперечная складка ладоней и др.) Основным симптомом является своеобразный мяукающий тембр плача ребенка, связанный с аномалией строения гортани.
Синдром Вольфа—Хиршхорна.
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорожденных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со складками, пучеглазие и колобома радужной оболочки (ее частичное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей.
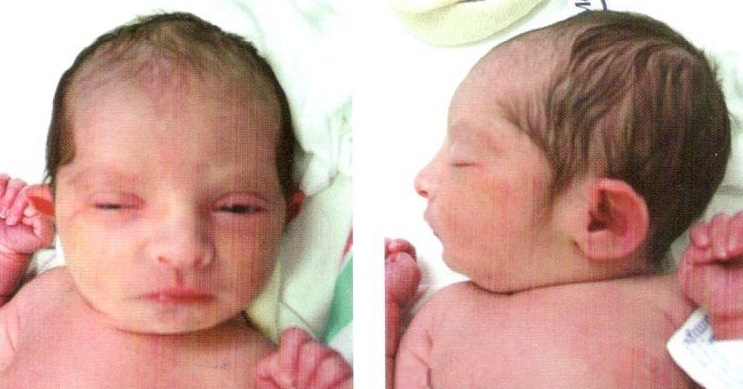
Синдром Патау - комплекс врожденных пороков развития черепа, лица, нервной системы, органов слуха, зрения, внутренних органов. В основе заболевания лежит наличие добавочной хромосомы в 13-й паре. Синдром описан в 1960 г. американским педиатром Патау (К. Patau).
Клиническая картина характеризуется микроцефалией, расщелиной лица, двусторонним расщеплением верхней губы, полным расщеплением неба, маленькими глазными яблоками либо полным их отсутствием, короткой шеей, маленькими деформированными низко расположенными ушами, полидактилией, дистрофическими изменениями ногтей и костного скелета. Отмечаются также пороки развития сердца, желудка, кишечника и других органов.
Синдром трисомии-Х впервые описан в 1959 г. Частота данной патологии составляет среди новорожденных 0,1%, а среди умственно отсталых — 0,6%. Большинство лиц женского пола с трисомией-Х выявляется среди больных психиатрических лечебниц. Клиническая картина характеризуется аномалиями развития скелета, внутренних органов, различными психическими проявлениями и интеллектуальной недостаточностью. Среди полиморфизма признаков трисомии-Х наиболее характерными являются: низкий рост, аномалии ушей, прикуса, высокое стояние твердого неба, короткие пальцы, искривленный мизинец, широкий промежуток между 1 и 2 пальцами на стопах, синдактилия, недоразвитие половых функций.
Умственная отсталость проявляется в виде легкой или умеренной степени. Характерны эмоциональные расстройства (вспыльчивость, агрессивность, неустойчивость настроения и немотивированные поступки). Девочки с синдромом трисомии-Х с трудом, но в большинстве случаев (легкая степень умственной отсталости) обучаются в массовых школах.
Синдром Эдвардса - наследственное заболевание, обусловленное, как правило, трисомией 18-й хромосомы и проявляющееся множественными пороками развития органов и систем. Синдром описан в 1960 г. американским педиатром Эдвардсом (J. Edwards).
Клиническая картина заболевания характеризуется задержкой психического развития, множественными аномалиями лица, костно-мышечной системы, черепа и головного мозга.
К хромосомным синдромам, помимо вышеописанных, относится большая группа так называемых семейных форм умственной отсталости, когда совершенно точно доказано наличие данной патологии у близких родственников.
Синдром Аперта (акроцефалосиндактилия) — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, экзофтальмом, деформацией зубов и синдактилиями. Синдром описан французским педиатром Апертом (Е. Apert) в 1906 г.
Синдром Крузона — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, преждевременным срастанием швов черепа, уменьшением мозгового вещества, экзофтальмом, вторичной атрофией зрительных нервов, прямоугольным расположением большого пальца к кисти. Впервые синдром описан французским врачом Крузоном (О. Crouson) в 1912 г.
Синдром Сьегрена—Ларссона - наследственное заболевание, которое сопровождается умственной отсталостью, парезами (снижением силы) конечностей и ухудшением зрения.
Синдром Берьесона—Форсмана—Лемана — синдром характеризующийся умственной отсталостью в сочетании с избыточным весом. Впервые описан американскими врачами Берьесоном (М. Berjeson) Форсманом (Н. Foreman) и Леманом (О. Lehman) в 1963 г. Клиническая картина заболевания проявляется выраженным ожирением и прогрессирующей умственной отсталостью. Ожирение носит не равномерный характер. Жир откладывается преимущественно на бедрах, груди и лице, что придает своеобразный вид такому больному (бочкообразная карликовая фигура с заплывшим лицом, большими ушами и узкими разрезами глаз). У больных часто отмечаются эпилептические припадки. Умственная отсталость колеблется от умеренной до тяжелой степени. Данная патология встречается только у лиц мужского пола, но носителями патологического гена являются женщины.
Синдром Прадера—Вилли — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гипогенитализмом, ожирением, резко выраженной мышечной гипотонией.
Синдром Книппеля—Фейля (синдром короткой шеи) — наследственное семейное заболевание, обусловленное врожденными аномалиями развития скелета и внутренних органов в сочетании с тяжелой степенью умственной отсталости. Клиника синдрома подробно описана французскими врачами Клиппелем Фейлем в 1912 г.
Аномалия развития характеризуется следующими проявлениями: короткой шеей как результат количественного уменьшения шейных позвонков, ограничением подвижности головы, расщеплением твердого неба, бочкообразной грудной клеткой, врожденными пороками сердца, добавочными долями или отсутствием отдельных долей легких, синдактилиями (сращение пальцев конечностей), глухотой вследствие заращения наружных слуховых проходов, сужением анального отверстия и многими другими симптомами. Интеллектуальная недостаточность является результатом тяжелой умственной отсталости
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии - биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки - метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Специализация центра
Отзывы пациентов о лечении
















































































































































Фотоальбом центра Cortex